


地址:上海市宝山区长江软件园
电话:191 2100 2160

微信联系我们
2023-11-15
来源:上海百沐生物科技有限公司
所属分类:单细胞测序100问
质量控制的第一步是从数据集中删除低质量的读数。当细胞检测到的基因数量较少、计数深度较低且线粒体计数较高时,细胞膜可能会破裂,这表明细胞正在死亡。由于这些细胞通常不是我们分析的主要目标,并且可能会扭曲我们的下游分析,因此我们在质量控制过程中将其去除。为了识别它们,我们定义了细胞质量控制(QC)阈值。细胞质控通常对以下三个质控协变量进行:
每个条形码的计数数量(计数深度)
每个条形码的基因数量
每个条形码的线粒体基因计数分数
在细胞 QC 中,这些协变量通过阈值过滤,因为它们可能对应于死亡细胞。正如所指出的,它们可能反映了膜破裂的细胞,其细胞质 mRNA 已泄漏,因此只有线粒体中的 mRNA 仍然存在。这些细胞可能会显示出较低的计数深度、很少检测到的基因以及较高比例的线粒体读数。然而,共同考虑三个 QC 协变量至关重要,否则可能会导致细胞信号的误解。例如,线粒体计数相对较高的细胞可能参与呼吸过程,不应被过滤掉。然而,计数低或高的细胞可能对应于静止细胞群或尺寸较大的细胞。因此,在对单个协变量做出阈值决策时,优选考虑多个协变量。一般来说,建议排除较少的细胞并尽可能允许,以避免过滤掉活细胞
群或小亚群。
仅对少数或小型数据集进行 QC 通常以手动方式执行,方法是查看不同 QC 协变量的分布并识别随后将被过滤的异常值。然而,随着数据集规模的增长,这项任务变得越来越耗时,可能值得考虑通过 MAD(中值绝对偏差)进行自动阈值处理。
在 QC 中,第一步是计算 QC 协变量或指标。我们使用 scanpy 函数来计算这些 sc.pp.calculate_qc_metrics,该函数还可以计算特定基因群体的计数比例。因此,我们定义了线粒体、核糖体和血红蛋白基因。值得注意的是,线粒体计数用前缀“mt-”或“MT-”注释,具体取决于数据集中考虑的物种。如前所述,本笔记本中使用的数据集是人骨髓,因此线粒体计数用前缀“MT-”注释。对于鼠标数据集,前缀通常是小写,即“mt-”。
# mitochondrial genes
adata.var["mt"] = adata.var_names.str.startswith("MT-")
# ribosomal genes
adata.var["ribo"] = adata.var_names.str.startswith(("RPS", "RPL"))
# hemoglobin genes.
adata.var["hb"] = adata.var_names.str.contains(("^HB[^(P)]"))
我们现在可以使用 scanpy 计算相应的 QC 指标。
sc.pp.calculate_qc_metrics(
adata, qc_vars=["mt", "ribo", "hb"], inplace=True, percent_top=[20], log1p=True
)
adata
AnnData object with n_obs × n_vars = 16934 × 36601
obs:'n_genes_by_counts','log1p_n_genes_by_counts','total_counts','log1p_total_counts','pct_counts_in_top_20_genes','total_counts_mt','log1p_total_counts_mt','pct_counts_mt','total_counts_ribo','log1p_total_counts_ribo','pct_counts_ribo','total_counts_hb', 'log1p_total_counts_hb', 'pct_counts_hb'
正如我们所看到的,该函数向.var和.obs添加了几个附加列。在这里重点介绍其中的一些,有关不同指标的更多信息可以在 scanpy 文档中找到:
n_genes_by_countsin.obs是细胞中计数呈阳性的基因数量。
total_counts是细胞的计数总数,这也可能称为库大小。
pct_counts_mt是线粒体细胞总数的比例。
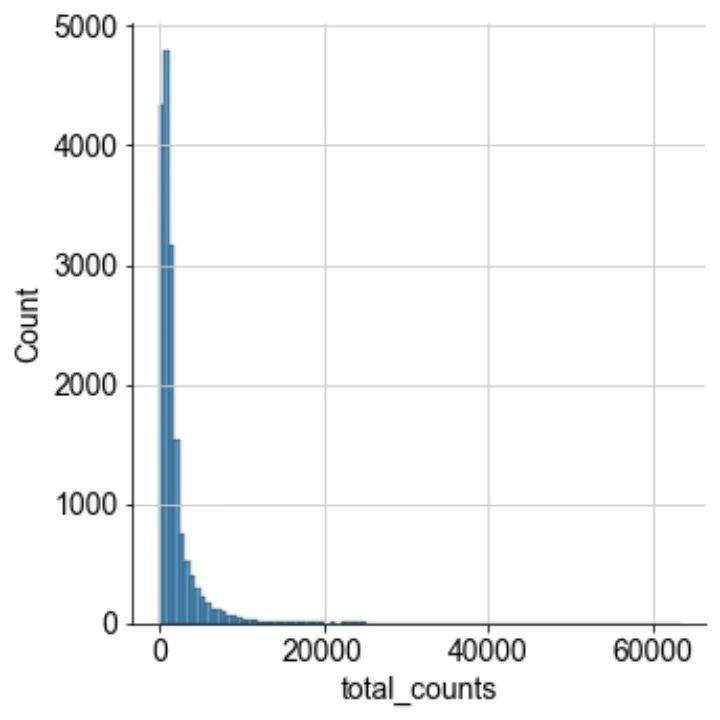
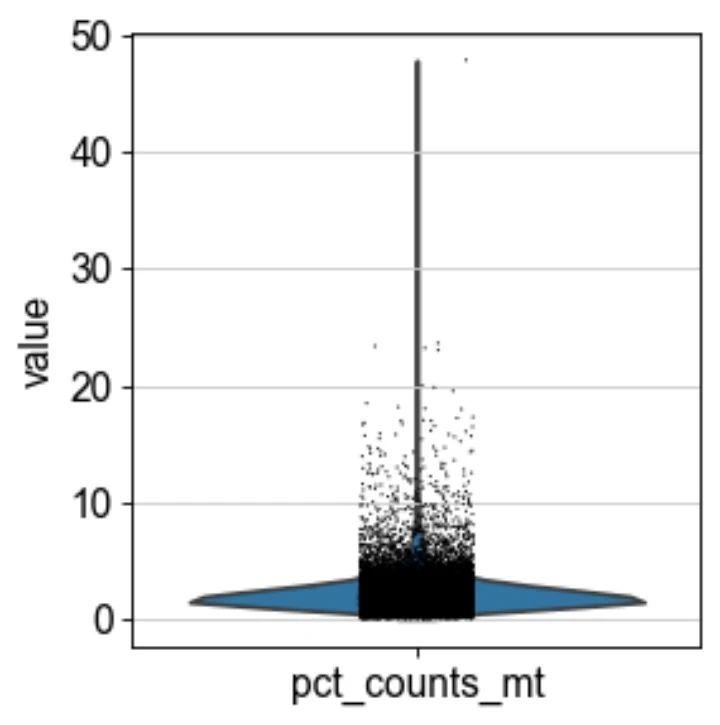
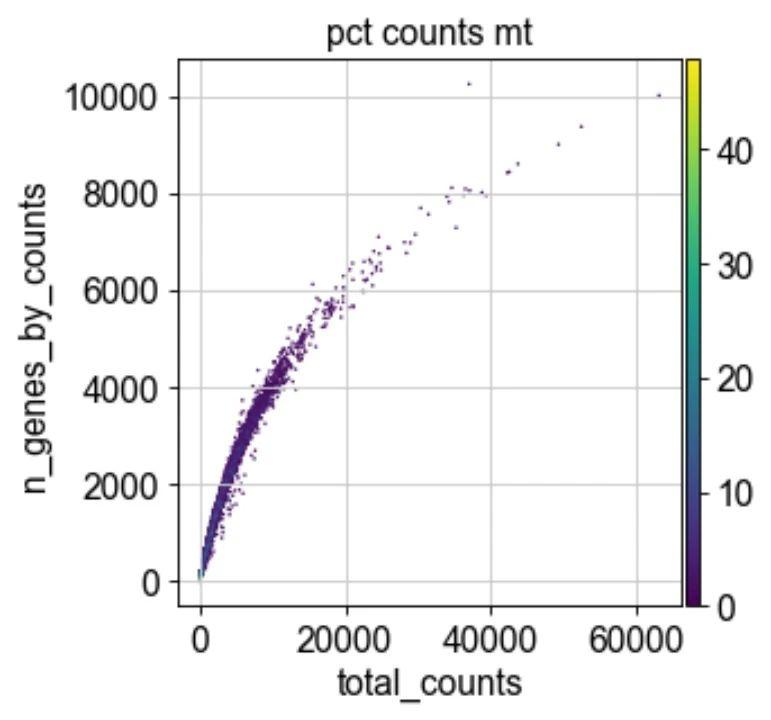
我们现在绘制三个样本的QC协变量 n_genes_by_counts, total_counts and pct_counts_mt,以评估各个细胞的捕获情况。
p1 = sns.displot(adata.obs["total_counts"], bins=100, kde=False)
# sc.pl.violin(adata, 'total_counts')
p2 = sc.pl.violin(adata, "pct_counts_mt")
p3 = sc.pl.scatter(adata, "total_counts", "n_genes_by_counts", color="pct_counts_mt")
... storing 'feature_types' as categorical
... storing 'genome' as categorical
图一
图二
图三
这些图表明,一些读数具有相对较高百分比的线粒体计数,通常与细胞降解相关。但由于每个细胞的计数数量足够高,并且大多数细胞的线粒体读数百分比低于20%,我们仍然可以处理数据。基于这些图,现在还可以定义用于过滤细胞的手动阈值。这里我们将展示基于 MAD的自动阈值和过滤的QC。
首先,我们定义一个函数。
def is_outlier(adata, metric: str, nmads: int):
M = adata.obs[metric]
outlier = (M < np.median(M) - nmads * median_abs_deviation(M)) | (
np.median(M) + nmads * median_abs_deviation(M) < M
)
return outlier
我们现在将此函数应用于log1p_total_counts、log1p_n_genes_by_counts和pct_counts_in_top_20_genesQC协变量,每个协变量的阈值为5 MADs。
adata.obs["outlier"] = (
is_outlier(adata, "log1p_total_counts", 5)
| is_outlier(adata, "log1p_n_genes_by_counts", 5)
| is_outlier(adata, "pct_counts_in_top_20_genes", 5)
)
adata.obs.outlier.value_counts()
False 16065
True 869
Name: outlier, dtype: int64
pct_counts_Mt使用3个MADs进行过滤。此外,线粒体计数百分比超过 8% 的细胞也会被滤除。
adata.obs["mt_outlier"] = is_outlier(adata, "pct_counts_mt", 3) | (
adata.obs["pct_counts_mt"] > 8
)
adata.obs.mt_outlier.value_counts()
False 15240
True 1694
Name: mt_outlier, dtype: int64
现在,我们根据这两个附加列来过滤 AnnData 对象。
print(f"Total number of cells: {adata.n_obs}")
adata = adata[(~adata.obs.outlier) & (~adata.obs.mt_outlier)].copy()
print(f"Number of cells after filtering of low quality cells: {adata.n_obs}")
Total number of cells: 16934
Number of cells after filtering of low quality cells: 14814
p1 = sc.pl.scatter(adata, "total_counts", "n_genes_by_counts", color="pct_counts_mt")
 在线咨询
在线咨询 微信咨询
微信咨询
 电话咨询
电话咨询
联系电话:191 2100 2160
 顶部
顶部